
sábado, 11 de abril de 2015
SÍNDROME DE PROTEUS
Es un trastorno congénito raro y esporádico que consiste en el crecimiento desproporcionado de múltiples tejidos, la mayor parte de origen mesodérmico, malformaciones vasculares y nervios de tejido conectivo o epidérmico. Las principales características de esta enfermedad hamartomatosa son: gigantismo parcial de las manos y los pies, hemihipertrofia, masas subcutáneas, nevó epidérmico y anormalidades óseas. Los pacientes con síndrome de Proteus tienen diversos y variados fenotipos debido a su patrón de distribución en mosaico.
SÍNDROME DE WAARDENBURG
Es una enfermedad hereditaria caracterizada por albinismo parcial (piel, cabello y ojos decolorados) y sordera neurosensorial. Se hereda como un rasgo autosómico dominante, lo que significa que es suficiente con el gen de sólo uno de los padres para que el niño resulte afectado.Existen cuatro tipos principales de este síndrome y los más comunes son el tipo I y el tipo II.
El tipo III (síndrome de Klein-Waardenburg) y el tipo IV (síndrome de Waandenburg-Shah) son menos comunes.
Los múltiples tipos de este síndrome resultan de las mutaciones que ocurren en diferentes genes. Todos los tipos comparten dos características dominantes: pérdida de la audición y cambios en la pigmentación (color) en la piel, el cabello y los ojos. Se puede presentar un mechón de pelo blanco en una cabeza de cabello normalmente oscuro. Los ojos pueden ser de un azul muy claro o de color diferente. Las personas afectadas con el tipo I casi siempre pueden tener una separación amplia de los ángulos internos de los ojos. La pérdida de la audición ocurre con más frecuencia en personas con el tipo II que con el tipo I de la enfermedad. Los tipos menos comunes de esta enfermedad pueden causar problemas con los brazos o los intestinos.

Los síntomas pueden abarcar:
- Labio leporino (infrecuente)
- Estreñimiento
- Sordera (más común en la enfermedad de tipo II)
- Ojos azules extremadamente pálidos u ojos de color diferente
- Piel, cabello y ojos de color claro (albinismo parcial)
- Dificultad para enderezar completamente las articulaciones
- Probable disminución leve de la capacidad intelectual
- Ojos ampliamente separados (en el tipo I)
- Mechón de pelo blanco o encanecimiento prematuro del cabello
SÍNDROME DE ARLEQUÍN
Característica
Se caracteriza por una aparición súbita de rubor y sudoración ante estímulos térmicos, emocionales o gustatorios. No obstante también pueden asociarse un síndrome de Holmes-Adie (pupila tónica y arreflexia), un síndrome de Ross (anhidrosis segmentaria, pupila tónica y arreflexia) o un síndrome de Horner contralateral a la zona de sudoración y rubor que se explicaría por una reacción compensatoria descrita en el anterior punto. En este último caso hablaríamos de signo del Arlequín, reservando el término de síndrome del Arlequín para cuando no existe una aparente afectación del simpático ocular (término también aplicado a un tipo de ictiosis congénita). Por último comentar que las alteraciones sudomotoras también pueden afectar a brazo y tronco lo cual implicaría una lesión proximal al ganglio estrellado.Por otro lado, y dadas las características anatómico-topográficas descritas, no es de extrañar que el síndrome del Arlequín se acompañe de un síndrome de Horner, ya sea clínicamente definido o puesto de manifiesto mediante los test farmacológicos oportunos con colirios.- Ectropión: protuberancia de los ojos hacia fuera.
- Eclabium: Igual que 1, pero en los labios.
- Membrana transparente y grasosa llamada colodión.
- Eritrodermia: 100% de la superficie corporal afectada.
Es por esto que el síndrome se ha descrito asociado a numerosas entidades clínicas (infartos bulbares, siringomielia, lesiones tumorales paravertebrales, en ápex pulmonar o torácicas) y como efecto deletéreo de procedimientos quirúrgicos o anestésicos (toracoscopia, simpatectomías para el tratamiento de hiperhidrosis, bloqueos epidurales torácicos o cateterismos de la yugular interna). Cuando no se encuentra la causa, se ha sugerido un origen del cuadro consistente en una compresión de la arteria radicular anterior por parte de la columna dorsal ante estímulos de torsión.
Se caracterizan clínicamente por escamas visibles, que adoptan diferentes patrones de distribución pudiendo ser localizadas o generalizadas e histológicamente (histología es la parte de la anatomía que estudia los tejidos que forman los seres vivos) por hiperqueratosis (hipertrofia, desarrollo exagerado, de la capa córnea de la piel), que suele estar asociada con algún grado de atrofia (disminución de volumen y peso de un órgano) de la epidermis. En algunas variantes de la enfermedad se puede observar paraqueratosis, queratosis pilosa y atrofia de las glándulas anejas. La gravedad de la ictiosis oscila desde una sequedad leve, aunque molesta, a una sequedad grave con descamación que puede llegar a ser desfigurante.

SÍNDROME DE PATAU
Síndrome congénito polimalformativo
grave, con una supervivencia que raramente
supera el año de vida, causado por la
existencia de tres copias del cromosoma 13
en el cariotipo. Al igual que otras trisomías humanas, la
mayoría de los casos de trisomía 13 se
deben a una no-disyunción cromosómica
durante la meiosis, principalmente en el
gameto materno. En estos embarazos la
edad materna y paterna media están algo
incrementadas (31.3 y 33.7 años respectivamente).
Aproximadamente un 20% de
casos se deben a traslocaciones, siendo la (13q14) la más frecuente. Sólo un 5% de
dichas traslocaciones es heredada de uno
de los progenitores. Los mosaicos representan
otro 5% de los casos de trisomía 13, en
los que el cuadro malformativo suele ser
menos grave.
La prevalencia de la trisomía 13 es de aproximadamente 1:12.000 nacidos vivos. La tasa de abortos espontáneos es elevada y representa alrededor del 1% del total de abortos espontáneos reconocidos. Existe un ligero exceso de casos del sexo femenino respecto al masculino.
Síntomas
- Labio leporino o paladar hendido
- Manos empuñadas (con los dedos externos sobre los dedos internos)
- Ojos muy juntos: los ojos pueden realmente fusionarse en uno
- Disminución del tono muscular
- Dedos adicionales en manos o pies
- Hernias
- Agujero, división o hendidura en el iris
- Orejas de implantación baja
- Discapacidad intelectual severo
- Defectos del cuero cabelludo (ausencia de piel)
- Convulsiones
- Pliegue palmar único
- Anomalías esqueléticas de las extremidades
- Ojos pequeños
- Cabeza pequeña
- Mandíbula inferior pequeña
- Testículo no descendido
La prevalencia de la trisomía 13 es de aproximadamente 1:12.000 nacidos vivos. La tasa de abortos espontáneos es elevada y representa alrededor del 1% del total de abortos espontáneos reconocidos. Existe un ligero exceso de casos del sexo femenino respecto al masculino.
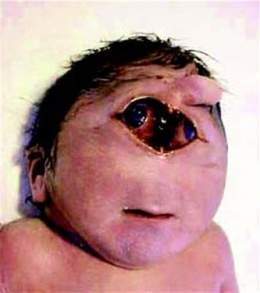
SÍNDROME DE DOWN
EL SÍNDROME DE DOWN (SD)
También llamado trisomía 21, es la causa mas frecuente de retraso mental identificable de origen genético. Se trata de una anomalía cromosómica que tiene una incidencia de 1 de cada 800 nacidos, y que aumenta con la edad materna. Es la cromosomopatía mas frecuente y mejor conocida.En el 95% de casos, el SD se produce por una trisomía del cromosoma 21 debido generalmente a la no disyunción meiótica en el óvulo. Aproximadamente un 4% se debe a una traslocación robertsoniana entre el cromosoma 21 y otro cromosoma acrocéntrico que normalmente es el 14 o el 22. Ocasionalmente puede encontrarse una traslocación entre dos cromosomas 21. Por último un 1% de los pacientes presentan un mosaico, con cariotipo normal y trisomía 21. No existen diferencias fenotípicas entre los diferentes tipos de SD. La realización del cariotipo es obligada para realizar un adecuado asesoramiento genético dado que el riesgo de recurrencia depende del cariotipo del paciente.
CLÍNICA
Los niños con SD se caracterizan por presentar una gran hipotonía e hiperlaxitud ligamentosa. Fenotípicamente presentan unos rasgos muy característicos. (figura 1) CABEZA y CUELLO: leve microcefalia con braquicefalia y occipital aplanado. El cuello es corto. CARA: los ojos son “almendrados”, y si el iris es azul suele observarse una pigmentación moteada, son las manchas de B r u s h f i e l d. Las hendiduras palpebrales siguen una dirección oblicua hacia arriba y afuera y presentan un pliegue de piel que cubre el ángulo interno y la carúncula del ojo (epicanto). La nariz es pequeña con la raíz nasal aplanada. La boca también es pequeña y la protusión lingual característica. Las orejas son pequeñas con un helix muy plegado y habitualmente con ausencia del lóbulo. El conducto auditivo puede ser muy estrecho. MANOS Y PIES: manos pequeñas y cuadradas con metacarpianos y falanges cortas (braquidactilia) y clinodactilia por hipoplasia de la falange media del 5º dedo. Puede observarse un surco palmar único. En el pie existe una hendidura entre el primer y segundo dedo con un aumento de la distancia entre los mismos (signo de la sandalia). GENITALES: el tamaño del pene es algo pequeño y el volumen testicular es menor que el de los niños de su edad, una criptorquídia es relativamente frecuente en estos individuos. PIEL y FANERAS: la piel es redundante en la región cervical sobretodo en el perí- odo fetál y neonatal. Puede observarselivedo reticularis (cutis marmorata) de predominio en extremidades inferiores. Con el tiempo la piel se vuelve seca e hiperestesiara. El retraso mental es constante en mayor o menor grado. DIAGNÓSTICO Las características fenotípicas del SD pueden no ser muy evidentes en el período neonatal inmediato. En este momento la gran hipotonía y el llanto característico, agudo y entrecortado, pueden ser la clave para el diagnóstico. Al poco tiempo se define el fenotipo característico, aunque cada uno tendrá sus propias peculiaridades. El diagnóstico definitivo vendrá dado por el estudio de los cromosomas. RIESGO DE RECURRENCIA El SD puede diagnosticarse prenatalmente realizando un estudio citogenético de vellosidades coriónicas o de líquido amnió- tico. El riesgo depende de la edad materna, pero también del cariotipo de los progenitores. (tabla I). En el caso que se trate de una trisomía 21, el riesgo de recurrencia para las mujeres de edad superior a los 30 años es el mismo que le da su edad. En las mujeres mas jóvenes es algo mas alto. En el caso de que exista una traslocación y alguno de los progenitores sea portador, no influye la edad materna, pero existe un riesgo mas alto de recurrencia si el portador de la traslocación es la madre. En el caso de que alguno de los padres tenga una traslocación Robertsoniana entre dos cromosomas 21 el riesgo de recurrencia es del 100% independientemente del sexo que lo transmita. Si ninguno de los progenitores es portador de una traslocación el riesgo de recurrencia es de alrededor de un 2-3% SEGUIMIENTO Los niños con SD deben seguir los controles periódicos (tabla II) y vacunas como cualquier otro niño de la misma edad, pero además se debe prestar especial atención a las complicaciones que pueden aparecer inherentes a su cromosomopatía.. Deben usarse gráficas de crecimiento específicas para el SD (www.growthcharts.com), y si existe un retraso pondoestatural muy marcadas nos puede orientar hacia la existencia de una patología cardíaca, endocrina o a una alteración nutricional. Un 30-60% de los SD presentarán una cardiopatía. Aunque clínicamente no se constate un soplo cardíaco, se deberá realizar un ecocardiograma en los primeros dos meses de vida. Si existe cardiopatía se deberá insistir a los padres en la necesidad de profilaxis antibiótica ante cualquier procedimiento que suponga un riesgo de endocarditis bacteriana (dental, nefrourológico...). No debe olvidarse el riesgo de desarrollar hipertensión pulmonar sobretodo en los niños con comunicación interventricular o con canal atrioventricular, que pueden estar asintomáticos en el primer año de vida.

También llamado trisomía 21, es la causa mas frecuente de retraso mental identificable de origen genético. Se trata de una anomalía cromosómica que tiene una incidencia de 1 de cada 800 nacidos, y que aumenta con la edad materna. Es la cromosomopatía mas frecuente y mejor conocida.En el 95% de casos, el SD se produce por una trisomía del cromosoma 21 debido generalmente a la no disyunción meiótica en el óvulo. Aproximadamente un 4% se debe a una traslocación robertsoniana entre el cromosoma 21 y otro cromosoma acrocéntrico que normalmente es el 14 o el 22. Ocasionalmente puede encontrarse una traslocación entre dos cromosomas 21. Por último un 1% de los pacientes presentan un mosaico, con cariotipo normal y trisomía 21. No existen diferencias fenotípicas entre los diferentes tipos de SD. La realización del cariotipo es obligada para realizar un adecuado asesoramiento genético dado que el riesgo de recurrencia depende del cariotipo del paciente.
CLÍNICA
Los niños con SD se caracterizan por presentar una gran hipotonía e hiperlaxitud ligamentosa. Fenotípicamente presentan unos rasgos muy característicos. (figura 1) CABEZA y CUELLO: leve microcefalia con braquicefalia y occipital aplanado. El cuello es corto. CARA: los ojos son “almendrados”, y si el iris es azul suele observarse una pigmentación moteada, son las manchas de B r u s h f i e l d. Las hendiduras palpebrales siguen una dirección oblicua hacia arriba y afuera y presentan un pliegue de piel que cubre el ángulo interno y la carúncula del ojo (epicanto). La nariz es pequeña con la raíz nasal aplanada. La boca también es pequeña y la protusión lingual característica. Las orejas son pequeñas con un helix muy plegado y habitualmente con ausencia del lóbulo. El conducto auditivo puede ser muy estrecho. MANOS Y PIES: manos pequeñas y cuadradas con metacarpianos y falanges cortas (braquidactilia) y clinodactilia por hipoplasia de la falange media del 5º dedo. Puede observarse un surco palmar único. En el pie existe una hendidura entre el primer y segundo dedo con un aumento de la distancia entre los mismos (signo de la sandalia). GENITALES: el tamaño del pene es algo pequeño y el volumen testicular es menor que el de los niños de su edad, una criptorquídia es relativamente frecuente en estos individuos. PIEL y FANERAS: la piel es redundante en la región cervical sobretodo en el perí- odo fetál y neonatal. Puede observarselivedo reticularis (cutis marmorata) de predominio en extremidades inferiores. Con el tiempo la piel se vuelve seca e hiperestesiara. El retraso mental es constante en mayor o menor grado. DIAGNÓSTICO Las características fenotípicas del SD pueden no ser muy evidentes en el período neonatal inmediato. En este momento la gran hipotonía y el llanto característico, agudo y entrecortado, pueden ser la clave para el diagnóstico. Al poco tiempo se define el fenotipo característico, aunque cada uno tendrá sus propias peculiaridades. El diagnóstico definitivo vendrá dado por el estudio de los cromosomas. RIESGO DE RECURRENCIA El SD puede diagnosticarse prenatalmente realizando un estudio citogenético de vellosidades coriónicas o de líquido amnió- tico. El riesgo depende de la edad materna, pero también del cariotipo de los progenitores. (tabla I). En el caso que se trate de una trisomía 21, el riesgo de recurrencia para las mujeres de edad superior a los 30 años es el mismo que le da su edad. En las mujeres mas jóvenes es algo mas alto. En el caso de que exista una traslocación y alguno de los progenitores sea portador, no influye la edad materna, pero existe un riesgo mas alto de recurrencia si el portador de la traslocación es la madre. En el caso de que alguno de los padres tenga una traslocación Robertsoniana entre dos cromosomas 21 el riesgo de recurrencia es del 100% independientemente del sexo que lo transmita. Si ninguno de los progenitores es portador de una traslocación el riesgo de recurrencia es de alrededor de un 2-3% SEGUIMIENTO Los niños con SD deben seguir los controles periódicos (tabla II) y vacunas como cualquier otro niño de la misma edad, pero además se debe prestar especial atención a las complicaciones que pueden aparecer inherentes a su cromosomopatía.. Deben usarse gráficas de crecimiento específicas para el SD (www.growthcharts.com), y si existe un retraso pondoestatural muy marcadas nos puede orientar hacia la existencia de una patología cardíaca, endocrina o a una alteración nutricional. Un 30-60% de los SD presentarán una cardiopatía. Aunque clínicamente no se constate un soplo cardíaco, se deberá realizar un ecocardiograma en los primeros dos meses de vida. Si existe cardiopatía se deberá insistir a los padres en la necesidad de profilaxis antibiótica ante cualquier procedimiento que suponga un riesgo de endocarditis bacteriana (dental, nefrourológico...). No debe olvidarse el riesgo de desarrollar hipertensión pulmonar sobretodo en los niños con comunicación interventricular o con canal atrioventricular, que pueden estar asintomáticos en el primer año de vida.
Suscribirse a:
Comentarios (Atom)